UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark one)
For the quarterly period ended
or
For the transition period from ________________ to __________________
Commission File Number:
(Exact name of registrant as specified in its charter)
|
(State or other jurisdiction of incorporation or organization) |
Not Applicable (I.R.S. Employer Identification No.) |
|
(Address of principal executive offices) (Zip Code) ( (Registrant’s telephone number, including area code)
|
|
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class |
Trading Symbol |
Name of each exchange on which registered |
|
|
|
The |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer ☐ |
Accelerated filer ☐ |
|
|
Smaller reporting company |
|
Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). YES
As of November 11, 2024, there were
DiaMedica Therapeutics Inc.
FORM 10-Q
September 30, 2024
TABLE OF CONTENTS
|
Description |
Page |
|
|
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS |
1 |
|
|
PART I. |
FINANCIAL INFORMATION |
|
|
Item 1. |
Financial Statements |
3 |
|
Item 2. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
14 |
|
Item 3. |
Quantitative and Qualitative Disclosures about Market Risk |
20 |
|
Item 4. |
Controls and Procedures |
20 |
|
PART II. |
OTHER INFORMATION |
|
|
Item 1. |
Legal Proceedings |
21 |
|
Item 1A. |
Risk Factors |
21 |
|
Item 2. |
Unregistered Sales of Equity Securities and Use of Proceeds |
22 |
|
Item 3. |
Defaults Upon Senior Securities |
22 |
|
Item 4. |
Mine Safety Disclosures |
22 |
|
Item 5. |
Other Information |
22 |
|
Item 6. |
Exhibits |
23 |
|
SIGNATURE PAGE |
24 |
|
This quarterly report on Form 10-Q contains certain forward-looking statements within the meaning of Section 27A of the United States Securities Act of 1933, as amended, and Section 21E of the United States Securities Exchange Act of 1934, as amended, that are subject to the safe harbor created by those sections. For more information, see “Cautionary Note Regarding Forward-Looking Statements.”
As used in this report, references to “DiaMedica,” the “Company,” “we,” “our” or “us,” unless the context otherwise requires, refer to DiaMedica Therapeutics Inc. and its subsidiaries, all of which are consolidated in DiaMedica’s condensed consolidated financial statements. References in this report to “common shares” mean our voting common shares, no par value per share.
We own various unregistered trademarks and service marks, including our corporate logo. Solely for convenience, the trademarks and trade names in this report are referred to without the ® and ™ symbols, but such references should not be construed as any indicator that the owner of such trademarks and trade names will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend the use or display of other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Statements in this report that are not descriptions of historical facts are forward-looking statements within the meaning of the United States Private Securities Litigation Reform Act of 1995 that are based on management’s current expectations and are subject to risks and uncertainties that could negatively affect our business, operating results, financial condition, prospects and share price. We have attempted to identify forward-looking statements by terminology including “anticipates,” “believes,” “can,” “continue,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “should,” “will,” “would,” the negative of these terms or other comparable terminology and the use of future dates.
The forward-looking statements in this report are subject to risks and uncertainties and include, among other things:
|
● |
our plans to develop, obtain regulatory approval for and commercialize our DM199 product candidate for the treatment of acute ischemic stroke (AIS) and preeclampsia (PE); |
|
● |
our expansion into PE, the ability of our physician collaborators to successfully conduct a Phase 2, proof-of-concept clinical trial of DM199 as a treatment for PE and our reliance on our physician collaborators; |
|
● |
our ability to conduct successful clinical testing of our DM199 product candidate for AIS and PE and meet certain anticipated or target dates with respect to our clinical studies,; |
|
● |
our ability to meet anticipated site activations, enrollment and interim analysis timing with respect to our Phase 2/3 ReMEDy2 clinical trial of DM199 for the treatment of AIS, especially in the light of slower than expected site activations and enrollment which we believe are due, in part, to hospital and medical facility staffing shortages; inclusion/exclusion criteria in the study protocol; concerns managing logistics and protocol compliance for participants discharged from the hospital to another hospital or an intermediate care facility; concerns regarding the prior clinically significant hypotension events and circumstances surrounding the clinical hold which was lifted in June 2023; and competition for research staff and trial subjects due to other pending stroke and neurological trials; |
|
● |
the success of the actions we are taking to mitigate the impact of the factors adversely affecting our ReMEDy2 trial site activations and enrollment rate, including significantly expanding our internal clinical team and bringing in-house certain trial activities, including study site identification, qualification and activation, clinical site monitoring, and overall program management; globally expanding the trial, and making certain changes to the study protocol; and risks associated with these mitigation actions; |
|
● |
uncertainties relating to regulatory applications and related filing and approval timelines, and the possibility of additional future adverse events associated with or unfavorable results from our ReMEDy2 trial or the Phase 2 investigator-sponsored PE trial; |
|
● |
the adaptive design of our ReMEDy2 trial, which is intended to enroll approximately 350 participants at up to 100 sites globally, and the possibility that these numbers and other aspects of the study could increase depending upon certain factors, including additional input from the United States Food and Drug Administration (FDA) and results of the interim analysis as determined by the independent data safety monitoring board; |
|
● |
our expectations regarding the perceived benefits of our DM199 product candidate over existing treatment options for AIS and PE; |
|
● |
our ability to partner with and generate revenue from biopharmaceutical or pharmaceutical partners to develop, obtain regulatory approval for and commercialize our DM199 product candidate for AIS and PE; |
|
● |
the potential size of the markets for our DM199 product candidate for AIS and PE and our or any future partner’s ability to serve those markets, the rate and degree of market acceptance of and ability to obtain coverage and adequate reimbursement for, our DM199 product candidate for AIS and PE both in the United States and internationally; |
|
● |
the success, cost and timing of our ReMEDy2 trial, as well as our reliance on our key executives, clinical personnel, advisors and third parties in connection with our ReMEDy2 trial; |
|
● |
our or any future partner’s ability to commercialize, market and manufacture DM199; |
|
● |
expectations regarding federal, state and foreign regulatory requirements and developments affecting our pending and future clinical trials and regulatory approvals of our DM199 product candidate for AIS and PE and future commercialization and manufacturing of such products if required regulatory approvals are obtained; |
|
● |
our estimates regarding expenses, market opportunity for our product candidates, future revenue, capital requirements, how long our current cash resources will last and need for additional financing; |
|
● |
our expectations regarding our ability to obtain and maintain intellectual property protection for our DM199 product candidate; |
|
● |
expectations regarding competition and our ability to obtain data exclusivity for our DM199 product candidate for AIS and PE; |
|
● |
our anticipated use of the net proceeds from our June 2024 private placement; and |
|
● |
our ability to obtain additional funding for our operations, including funding necessary to complete our current clinical trials and obtain regulatory approvals for our DM199 product candidate for AIS and/or PE. |
These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described under “Part I. Item 1A. Risk Factors” in our annual report on Form 10-K for the fiscal year ended December 31, 2023, in this quarterly report on Form 10-Q for the quarterly period ended September 30, 2024 and those described above and elsewhere in this report. Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this report may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements. Forward-looking statements should not be relied upon as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. Except as required by law, including the securities laws of the United States, we do not intend to update any forward-looking statements to conform these statements to actual results or to changes in our expectations.
PART I - FINANCIAL INFORMATION
|
ITEM 1. |
FINANCIAL STATEMENTS |
DiaMedica Therapeutics Inc.
Condensed Consolidated Balance Sheets
(In thousands, except share amounts)
|
September 30, 2024 |
December 31, 2023 |
|||||||
|
(unaudited) |
||||||||
|
ASSETS |
||||||||
|
Current assets: |
||||||||
|
Cash and cash equivalents |
$ | $ | ||||||
|
Marketable securities |
||||||||
|
Amounts receivable |
||||||||
|
Prepaid expenses and other assets |
||||||||
|
Total current assets |
||||||||
|
Non-current assets: |
||||||||
|
Deposits |
||||||||
|
Operating lease right-of-use asset, net |
||||||||
|
Property and equipment, net |
||||||||
|
Total non-current assets |
||||||||
|
Total assets |
$ | $ | ||||||
|
LIABILITIES AND EQUITY |
||||||||
|
Current liabilities: |
||||||||
|
Accounts payable |
$ | $ | ||||||
|
Accrued liabilities |
||||||||
|
Operating lease obligation |
||||||||
|
Finance lease obligation |
||||||||
|
Total current liabilities |
||||||||
|
Non-current liabilities: |
||||||||
|
Operating lease obligation |
||||||||
|
Finance lease obligation |
||||||||
|
Total non-current liabilities |
||||||||
|
Shareholders’ equity: |
||||||||
|
Common shares, par value; authorized; |
||||||||
|
Paid-in capital |
||||||||
|
Accumulated other comprehensive income |
||||||||
|
Accumulated deficit |
( |
) | ( |
) | ||||
|
Total shareholders’ equity |
||||||||
|
Total liabilities and shareholders’ equity |
$ | $ | ||||||
See accompanying notes to the condensed consolidated financial statements.
DiaMedica Therapeutics Inc.
Condensed Consolidated Statements of Operations and Comprehensive Loss
(In thousands, except share and per share amounts)
(Unaudited)
|
Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
|
2024 |
2023 |
2024 |
2023 |
|||||||||||||
|
Operating expenses: |
||||||||||||||||
|
Research and development |
$ | $ | $ | $ | ||||||||||||
|
General and administrative |
||||||||||||||||
|
Operating loss |
( |
) |
( |
) |
( |
) | ( |
) | ||||||||
|
Other income: |
||||||||||||||||
|
Other income, net |
||||||||||||||||
|
Loss before income tax expense |
( |
) | ( |
) | ( |
) | ( |
) | ||||||||
|
Income tax expense |
( |
) |
( |
) |
( |
) | ( |
) | ||||||||
|
Net loss |
( |
) |
( |
) |
( |
) | ( |
) | ||||||||
|
Other comprehensive gain (loss) |
||||||||||||||||
|
Unrealized gain (loss) on marketable securities |
( |
) | ( |
) | ||||||||||||
|
Net loss and comprehensive loss |
$ | ( |
) |
$ | ( |
) |
$ | ( |
) | $ | ( |
) | ||||
|
Basic and diluted net loss per share |
$ | ( |
) |
$ | ( |
) |
$ | ( |
) | $ | ( |
) | ||||
|
Weighted average shares outstanding – basic and diluted |
||||||||||||||||
See accompanying notes to the condensed consolidated financial statements.
DiaMedica Therapeutics Inc.
Condensed Consolidated Statements of Shareholders’ Equity
For the Nine Months Ended September 30, 2024 and 2023
(In thousands, except share amounts)
(Unaudited)
|
Common Shares |
Paid-In Capital |
Accumulated Other Comprehensive Gain (Loss) |
Accumulated Deficit |
Total Shareholders’ Equity |
||||||||||||||||
|
Balances at December 31, 2023 |
$ | $ | $ | ( |
) | $ | ||||||||||||||
|
Issuance of common shares upon the vesting and settlement of restricted stock units |
||||||||||||||||||||
|
Share-based compensation expense |
— | |||||||||||||||||||
|
Unrealized loss on marketable securities |
— | ( |
) | ( |
) | |||||||||||||||
|
Net loss |
— | ( |
) | ( |
) | |||||||||||||||
|
Balances at March 31, 2024 |
$ | $ | ( |
) | $ | ( |
) | $ | ||||||||||||
|
Issuance of common shares net of offering costs of $ |
||||||||||||||||||||
|
Issuance of common shares upon the vesting and settlement of restricted stock units |
||||||||||||||||||||
|
Issuance of common shares upon the exercise of stock options |
||||||||||||||||||||
|
Share-based compensation expense |
— | |||||||||||||||||||
|
Unrealized loss on marketable securities |
— | ( |
) | ( |
) | |||||||||||||||
|
Net loss |
— | ( |
) | ( |
) | |||||||||||||||
|
Balances at June 30, 2024 |
$ | $ | ( |
) | $ | ( |
) | $ | ||||||||||||
|
Issuance of common shares upon the vesting of and settlement of restricted stock units |
||||||||||||||||||||
|
Issuance of common shares upon the exercise of stock options |
||||||||||||||||||||
|
Share-based compensation expense |
||||||||||||||||||||
|
Unrealized gain on marketable securities |
— | |||||||||||||||||||
|
Net loss |
— | ( |
) | ( |
) | |||||||||||||||
|
Balances at September 30, 2024 |
$ | $ | $ | ( |
) | $ | ||||||||||||||
|
Common Shares |
Paid-In Capital |
Accumulated Other Comprehensive Gain (Loss) |
Accumulated Deficit |
Total Shareholders’ Equity |
||||||||||||||||
|
Balances at December 31, 2022 |
$ | $ | ( |
) | $ | ( |
) | $ | ||||||||||||
|
Issuance of common shares in settlement of deferred stock units |
— | — | — | — | ||||||||||||||||
|
Issuance of common shares upon the vesting and settlement of restricted stock units |
||||||||||||||||||||
|
Share-based compensation expense |
— | |||||||||||||||||||
|
Unrealized gain on marketable securities |
— | |||||||||||||||||||
|
Net loss |
— | ( |
) | ( |
) | |||||||||||||||
|
Balances at March 31, 2023 |
$ | $ | ( |
) | $ | ( |
) | $ | ||||||||||||
|
Issuance of common shares net of offering costs of $ |
||||||||||||||||||||
|
Issuance of common shares upon the vesting and settlement of restricted stock units |
||||||||||||||||||||
|
Share-based compensation expense |
— | |||||||||||||||||||
|
Unrealized loss on marketable securities |
— | ( |
) | ( |
) | |||||||||||||||
|
Net loss |
— | ( |
) | ( |
) | |||||||||||||||
|
Balances at June 30, 2023 |
$ | $ | ( |
) | $ | ( |
) | $ | ||||||||||||
|
Offering costs on previously issued common shares |
— | ( |
) | ( |
) | |||||||||||||||
|
Issuance of common shares upon the vesting and settlement of restricted stock units |
||||||||||||||||||||
|
Share-based compensation expense |
||||||||||||||||||||
|
Unrealized loss on marketable securities |
— | ( |
) | ( |
) | |||||||||||||||
|
Net loss |
— | ( |
) | ( |
) | |||||||||||||||
|
Balances at September 30, 2023 |
$ | $ | ( |
) | $ | ( |
) | $ | ||||||||||||
See accompanying notes to the condensed consolidated financial statements.
DiaMedica Therapeutics Inc.
Condensed Consolidated Statements of Cash Flows
(In thousands)
(Unaudited)
|
Nine Months Ended September 30, |
||||||||
|
2024 |
2023 |
|||||||
|
Cash flows from operating activities: |
||||||||
|
Net loss |
$ | ( |
) |
$ | ( |
) |
||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
|
Share-based compensation |
||||||||
|
Amortization of discount on marketable securities |
( |
) |
( |
) | ||||
|
Non-cash lease expense |
||||||||
|
Depreciation |
||||||||
|
Changes in operating assets and liabilities: |
||||||||
|
Amounts receivable |
( |
) | ||||||
|
Prepaid expenses and other assets |
( |
) | ||||||
|
Deposits |
( |
) | ||||||
|
Accounts payable |
||||||||
|
Accrued liabilities |
( |
) | ||||||
|
Net cash used in operating activities |
( |
) |
( |
) |
||||
|
Cash flows from investing activities: |
||||||||
|
Purchase of marketable securities |
( |
) |
( |
) |
||||
|
Maturities of marketable securities |
||||||||
|
Purchases of property and equipment |
( |
) |
( |
) |
||||
|
Net cash provided by (used in) investing activities |
( |
) | ||||||
|
Cash flows from financing activities: |
||||||||
|
Proceeds from issuance of common shares, net of offering costs |
||||||||
|
Proceed from the exercise of stock options |
||||||||
|
Principal payments on finance lease obligation |
( |
) |
( |
) |
||||
|
Net cash provided by financing activities |
||||||||
|
Net decrease in cash and cash equivalents |
( |
) | ( |
) | ||||
|
Cash and cash equivalents at beginning of period |
||||||||
|
Cash and cash equivalents at end of period |
$ | $ | ||||||
|
Supplemental disclosure of non-cash transactions: |
||||||||
|
Assets acquired under financing lease |
$ | $ | ||||||
|
Cash paid for income taxes |
$ | $ | ||||||
See accompanying notes to the condensed consolidated financial statements.
DiaMedica Therapeutics Inc.
Notes to the Condensed Consolidated Financial Statements
(Unaudited)
1. Business
DiaMedica Therapeutics Inc. and its wholly owned subsidiaries, DiaMedica USA Inc. and DiaMedica Australia Pty Ltd. (collectively, we, us, our, DiaMedica and the Company), exist for the primary purpose of advancing the clinical and commercial development of our proprietary recombinant KLK1 protein called DM199, for the treatment of neurological and cardio-renal diseases. Currently, our primary focus is on developing DM199, a recombinant form of the human tissue kallikrein-1 (KLK1) protein, for the treatment of acute ischemic stroke (AIS) and preeclampsia (PE). Our parent company is governed under British Columbia’s Business Corporations Act, and our common shares are publicly traded on The Nasdaq Capital Market under the symbol “DMAC.”
2. Risks and Uncertainties
DiaMedica operates in a highly regulated and competitive environment. The development, manufacturing and marketing of pharmaceutical products require approval from, and are subject to ongoing oversight by, the United States Food and Drug Administration (FDA) in the United States, the European Medicines Agency (EMA) in the European Union and comparable agencies in other countries. We are in the clinical stage of development of our initial product candidate, DM199, for the treatment of AIS and PE. We have not completed the development of any product candidate and do not generate any revenues from the commercial sale of any product candidate. DM199 requires significant additional clinical testing and investment prior to seeking marketing approval and is not expected to be commercially available for at least three to four years, if at all.
We are currently conducting our ReMEDy2 clinical trial of DM199 for the treatment of AIS. Our ReMEDy2 clinical trial is a Phase 2/3, adaptive design, randomized, double-blind, placebo-controlled trial intended to enroll approximately 350 patients at up to 100 sites globally. Prior to the clinical hold of our ReMEDy2 trial, announced in July 2022 and fully lifted in June 2023, we had experienced and are now continuing to experience slower than expected site activations and enrollment in our ReMEDy2 trial. We believe these conditions may be due to hospital and medical facility staffing shortages; inclusion/exclusion criteria in the study protocol; concerns managing logistics and protocol compliance for participants discharged from the hospital to another hospital or an intermediate care facility; concerns regarding the prior clinically significant hypotension events and circumstances surrounding the previous clinical hold; and competition for research staff and trial subjects due to other pending stroke and neurological trials. We continue to reach out to current and potential study sites to understand the specific issues at each study site. In an effort to mitigate the impact of these factors, we have significantly expanded our internal clinical team and have brought in-house certain trial activities, including site identification, qualification and activation, clinical site monitoring and overall program management. In addition, we made the decision to globally expand the trial and to this end we have submitted or are in the process of preparing regulatory filings and identifying and engaging study sites in the countries of Canada, Australia and Georgia. We also recently revised the study protocol to widen the inclusion criteria and reduce the burden on participants and sites. We continue to work closely with our contract research organizations and other advisors to develop procedures to support both U.S. and global study sites and potential participants as needed. We intend to continue to monitor the results of these efforts and, if necessary, implement additional actions to mitigate the impact of these factors on our ReMEDy2 trial; however, no assurances can be provided as to the success of these mitigation actions and if or when these issues will resolve. The failure to resolve these issues will result in delays in our ReMEDy2 trial.
On October 9, 2024, we announced the receipt of regulatory approval from the South African Health Products Regulatory Authority (SAHPRA) to initiate an investigator-sponsored study of DM199 in PE. We are financially supporting the conduct of a Phase 2 open-label, single center, single-arm, safety and pharmacodynamic, proof-of-concept, investigator-sponsored study of DM199 for the treatment of PE at the Tygerberg Hospital, Cape Town, South Africa. Up to 90 women with PE and potentially an additional 30 subjects with fetal growth restriction may be evaluated. The first subject was enrolled in the fourth quarter of 2024. Part 1A topline study results are intended to demonstrate initial proof-of-concept including whether DM199 is safe, lowers blood pressure and dilates intrauterine arteries to increase placental blood flow. These results are expected in the first half of 2025.
Our future success is dependent upon the success of our development efforts, our ability to demonstrate clinical progress for our DM199 product candidate in the United States or other markets, our ability, or the ability of any future partner, to obtain required governmental approvals of our product candidate, our ability to license or market and sell our DM199 product candidate and our ability to obtain additional financing to fund these efforts.
As of September 30, 2024, we have incurred losses of $
Our principal source of cash has been net proceeds from the issuance of equity securities. Although we have previously been successful in obtaining financing through equity securities offerings, there is no assurance that we will be able to do so in the future. This is particularly true if our clinical data are not positive or if economic and market conditions deteriorate.
We expect that we will need substantial additional capital to further our research and development activities, complete the required clinical studies, regulatory activities and manufacturing development for our product candidate, DM199, or any future product candidates, to a point where they may be licensed or commercially sold. We expect our current cash, cash equivalents and marketable securities are sufficient to continue our ReMEDy2 trial, support the Phase 2 PE trial and otherwise fund our planned operations for at least the next 12 months from the date of issuance of these condensed consolidated financial statements. The amount and timing of our future funding requirements will depend on many factors, including timing and results of our ongoing development efforts, including our current ReMEDy2 trial and the rate of site activation and participant enrollment in the study, the Phase 2 PE trial, the potential expansion of our current development programs, the effects of ongoing site staffing shortages and other factors on our clinical trials and our operating expenses. We may require significant additional funds earlier than we currently expect and there is no assurance that we will not need or seek additional funding prior to such time, especially if market conditions for raising capital are favorable.
3. Summary of Significant Accounting Policies
Interim financial statements
We have prepared the accompanying condensed consolidated financial statements in accordance with accounting principles generally accepted in the United States (US GAAP) for interim financial information and with the instructions to Form 10-Q and Regulation S-X of the Securities and Exchange Commission (SEC). Accordingly, they do not include all of the information and footnotes required by US GAAP for complete financial statements. These condensed consolidated financial statements reflect all adjustments consisting of normal recurring accruals which, in the opinion of management, are necessary to present fairly our condensed consolidated financial position, condensed consolidated results of operations, condensed consolidated statement of shareholders’ equity and condensed consolidated cash flows for the periods and as of the dates presented. Our fiscal year ends on December 31. The condensed consolidated balance sheet as of December 31, 2023 was derived from our audited consolidated financial statements. These condensed consolidated financial statements should be read in conjunction with our annual consolidated financial statements and the notes thereto. The nature of our business is such that the results of any interim period may not be indicative of the results to be expected for the entire year.
Cash and cash equivalents
The Company considers all bank deposits, including money market funds and other investments, purchased with an original maturity to the Company of three months or less, to be cash and cash equivalents. The carrying amount of our cash equivalents approximates fair value due to the short maturity of the investments.
Marketable securities
The Company’s marketable securities may consist of obligations of the United States government and its agencies, bank certificates of deposit and/or investment grade corporate obligations, which are classified as available-for-sale. Marketable securities which mature within 12 months from their purchase date are included in current assets. Securities are generally valued based on market prices for similar assets using third party certified pricing sources and are carried at fair value. The amortized cost of marketable securities is adjusted for amortization of premiums or accretion of discounts to maturity. Such amortization or accretion is included in interest income. Realized gains and losses, if any, are calculated on the specific identification method. Interest income is included in other income in the condensed consolidated statements of operations.
We conduct periodic reviews to identify and evaluate each available-for-sale debt security that is in an unrealized loss position in order to determine whether an other-than-temporary impairment exists. An unrealized loss exists when the current fair value of an individual security is less than its amortized cost basis. Declines in fair value considered to be temporary and caused by noncredit-related factors of the issuer, are recorded in accumulated other comprehensive loss, which is a separate component of shareholders’ equity. Declines in fair value that are other than temporary or caused by credit-related factors of the issuer, are recorded within earnings as an impairment loss. There were no other-than-temporary unrealized losses as of September 30, 2024.
Fair value measurements
Under the authoritative guidance for fair value measurements, fair value is defined as the exit price, or the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants as of the measurement date. The authoritative guidance also establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs market participants would use in valuing the asset or liability developed based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the factors market participants would use in valuing the asset or liability developed based upon the best information available in the circumstances. The categorization of financial assets and financial liabilities within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement.
The hierarchy is broken down into three levels defined as follows:
Level 1 Inputs — quoted prices in active markets for identical assets and liabilities
Level 2 Inputs — observable inputs other than quoted prices in active markets for identical assets and liabilities
Level 3 Inputs — unobservable inputs
As of September 30, 2024, the Company believes that the carrying amounts of its other financial instruments, including amounts receivable, accounts payable and accrued liabilities, approximate their fair value due to the short-term maturities of these instruments. See Note 4, titled “Marketable Securities” for additional information.
4. Marketable Securities
The available-for-sale marketable securities are primarily comprised of investments in commercial paper, corporate bonds and government securities and consist of the following, measured at fair value on a recurring basis (in thousands):
|
Fair Value Measurements Using Inputs Considered as of: |
||||||||||||||||||||||||||||||||
| September 30, 2024 |
December 31, 2023 |
|||||||||||||||||||||||||||||||
|
Total |
Level 1 |
Level 2 |
Level 3 |
Total |
Level 1 |
Level 2 |
Level 3 |
|||||||||||||||||||||||||
|
Commercial paper and corporate bonds |
$ | $ | $ | $ | $ | $ | $ | $ | ||||||||||||||||||||||||
|
Government securities |
||||||||||||||||||||||||||||||||
|
Total |
$ | $ | $ | $ | $ | $ | $ | $ | ||||||||||||||||||||||||
Maturities of individual securities are less than one year, and the amortized cost of all securities approximated fair value as of September 30, 2024 and December 31, 2023. Accrued interest receivable on marketable securities is included in amounts receivable and was $
There were no transfers of assets between Level 1 and Level 2 of the fair value measurement hierarchy during the nine months ended September 30, 2024.
5. Amounts Receivable
Amounts receivable consisted primarily of accrued interest receivable on marketable securities of $
6. Deposits
We periodically advance funds to vendors engaged to support the performance of our clinical trials and related activities. The funds advanced are held, interest free, for varying periods of time and are generally recovered by the Company through application against final study/project invoices or refunded upon completion of services to be provided. Deposits are non-current when their expected recovery is not within the next twelve months.
7. Property and Equipment
Property and equipment, net, consisted of the following (in thousands):
|
September 30, 2024 |
December 31, 2023 |
|||||||
|
Furniture and equipment |
$ | $ | ||||||
|
Computer equipment |
||||||||
|
Leasehold improvements |
||||||||
|
Less accumulated depreciation |
( |
) | ( |
) | ||||
|
Property and equipment, net |
$ | $ | ||||||
8. Accrued Liabilities
Accrued liabilities consisted of the following (in thousands):
|
September 30, 2024 |
December 31, 2023 |
|||||||
|
Clinical trial costs |
$ | $ | ||||||
|
Research and development services |
||||||||
|
Compensation |
||||||||
|
Professional services fees |
||||||||
|
Other |
||||||||
|
Total accrued liabilities |
$ | $ | ||||||
9. Operating Lease
Office lease
Our operating lease costs were $
Maturities of our operating lease obligation are as follows as of September 30, 2024 (in thousands):
|
2024 |
$ | |||
|
2025 |
||||
|
2026 |
||||
|
2027 |
||||
|
2028 |
||||
|
Total lease payments |
$ | |||
|
Less interest portion |
( |
) | ||
|
Present value of operating lease obligation |
$ |
10. Shareholders’ Equity
Authorized capital shares
DiaMedica has authorized share capital of an unlimited number of voting common shares, and the shares do not have a stated par value. Common shareholders are entitled to receive dividends as declared by the Company, if any, and are entitled to one vote per share at the Company’s annual general meeting and any extraordinary or special general meeting.
Equity issued during the nine months ended September 30, 2024
On June 25, 2024, we entered into securities purchase agreements with accredited investors, pursuant to which we agreed to issue and sell an aggregate
In connection with the June 2024 private placement, we entered into a registration rights agreement (Registration Rights Agreement) with the investors pursuant to which we agreed to file with the United States Securities and Exchange Commission (SEC) a registration statement registering the resale of the shares sold in the June 2024 private placement (Resale Registration Statement). The Resale Registration Statement was filed with the SEC on July 10, 2024 and declared effective by the SEC on July 18, 2024. Under the terms of the Registration Rights Agreement, we agreed to keep the Resale Registration Statement effective at all times until the shares are no longer considered “Registrable Securities” under the Registration Rights Agreement and if we fail to keep the Resale Registration Statement effective, subject to certain permitted exceptions, we will be required to pay liquidated damages to the investors in an amount of up to 10% of the invested capital, excluding interest. We also agreed, among other things, to indemnify the selling holders under the Resale Registration Statement from certain liabilities and to pay all fees and expenses incident to our performance of or compliance with the Registration Rights Agreement.
During the nine months ended September 30, 2024,
Equity issued during the nine months ended September 30, 2023
On April 10, 2023, in conjunction with his appointment as Chief Business Officer of DiaMedica, Mr. David Wambeke purchased
On June 21, 2023, we entered into securities purchase agreements with accredited investors, pursuant to which we agreed to issue and sell an aggregate
In connection with the June 2023 private placement, we entered into a registration rights agreement (Registration Rights Agreement) with the investors pursuant to which we agreed to file with the United States Securities and Exchange Commission (SEC) a registration statement registering the resale of the shares sold in the June 2023 private placement (Resale Registration Statement). The Resale Registration Statement was filed with the SEC on June 30, 2023 and declared effective by the SEC on July 7, 2023. Under the terms of the Registration Rights Agreement, we agreed to keep the Resale Registration Statement effective at all times until the shares are no longer considered “Registrable Securities” under the Registration Rights Agreement and if we fail to keep the Resale Registration Statement effective, subject to certain permitted exceptions, we will be required to pay liquidated damages to the investors in an amount of up to 10% of the invested capital, excluding interest. We also agreed, among other things, to indemnify the selling holders under the Resale Registration Statement from certain liabilities and to pay all fees and expenses incident to our performance of or compliance with the Registration Rights Agreement.
During the nine months ended September 30, 2023,
Shares reserved
Common shares reserved for future issuance are as follows:
|
September 30, 2024 |
||||
|
Common shares issuable upon exercise of employee and non-employee stock options |
||||
|
Common shares issuable upon settlement of deferred stock units |
||||
|
Common shares issuable upon vesting and settlement of restricted stock units |
||||
|
Shares available for grant under the Amended and Restated 2019 Omnibus Incentive Plan |
||||
|
Shares available for grant under the 2021 Employment Inducement Incentive Plan |
||||
|
Total |
||||
11. Net Loss Per Share
We compute net loss per share by dividing our net loss (the numerator) by the weighted-average number of common shares outstanding (the denominator) during the period. Shares issued during the period and shares reacquired during the period, if any, are weighted for the portion of the period that they were outstanding. The computation of diluted earnings per share, or EPS, is similar to the computation of basic EPS except that the denominator is increased to include the number of additional common shares that would have been outstanding if the dilutive potential common shares had been issued. Our diluted EPS is the same as basic EPS due to common equivalent shares being excluded from the calculation, as their effect is anti-dilutive.
The following table summarizes our calculation of net loss per common share for the periods presented (in thousands, except share and per share data):
|
Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
|
2024 |
2023 |
2024 |
2023 |
|||||||||||||
|
Net loss |
$ | ( |
) |
$ | ( |
) |
$ | ( |
) | $ | ( |
) | ||||
|
Weighted average shares outstanding—basic and diluted |
||||||||||||||||
|
Basic and diluted net loss per share |
$ | ( |
) |
$ | ( |
) |
$ | ( |
) | $ | ( |
) | ||||
The following outstanding potential common shares were not included in the diluted net loss per share calculations as their effects were not dilutive:
|
Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
|
2024 |
2023 |
2024 |
2023 |
|||||||||||||
|
Employee and non-employee stock options |
||||||||||||||||
|
Common shares issuable under common share purchase warrants |
||||||||||||||||
|
Common shares issuable upon settlement of deferred stock units |
||||||||||||||||
|
Common shares issuable upon vesting and settlement of restricted stock units |
||||||||||||||||
12. Share-Based Compensation
Amended and Restated 2019 Omnibus Incentive Plan
The DiaMedica Therapeutics Inc. Amended and Restated 2019 Omnibus Incentive Plan (as amended from time to time, the 2019 Plan) was adopted by the Board of Directors (Board) on March 14, 2019 and approved by our shareholders at our 2019 Annual General Meeting of Shareholders held on May 22, 2019. Subsequent amendments to the plan, comprised principally of increasing the authorized shares under the plan, were approved by our shareholders at our 2022 and 2024 Annual General Meetings of Shareholders.
The 2019 Plan permits the Board, or a committee or subcommittee thereof, to grant to the Company’s eligible employees, non-employee directors and certain consultants non-statutory and incentive stock options, stock appreciation rights, restricted stock awards, restricted stock units (RSUs), deferred stock units (DSUs), performance awards, non-employee director awards and other share-based awards. We grant options to purchase common shares under the 2019 Plan at no less than the fair market value of the underlying common shares as of the date of grant. Options granted to employees and non-employee directors have a maximum term of years and generally vest over to years. Options granted to non-employees have a maximum term of years and generally vest over year. Subject to adjustment as provided in the 2019 Plan, the maximum number of the Company’s common shares authorized for issuance under the 2019 Plan is
2021 Employment Inducement Incentive Plan
On December 3, 2021, the Board adopted the DiaMedica Therapeutics Inc. 2021 Employment Inducement Incentive Plan (Inducement Plan) to facilitate the granting of equity awards as an inducement material to new employees joining the Company. The Inducement Plan was adopted without shareholder approval pursuant to Nasdaq Listing Rule 5635(c)(4) and is administered by the Compensation Committee of the Board of Directors. The Board reserved
Prior Stock Option Plan
The DiaMedica Therapeutics Inc. Stock Option Plan, Amended and Restated November 6, 2018 (Prior Plan), was terminated by the Board of Directors in conjunction with the shareholder approval of the 2019 Plan. Awards outstanding under the Prior Plan remain outstanding in accordance with and pursuant to the terms thereof. Options granted under the Prior Plan have terms similar to those used under the 2019 Plan. As of September 30, 2024, options to purchase an aggregate of
Prior Deferred Stock Unit Plan
The DiaMedica Therapeutics Inc. Amended and Restated Deferred Stock Unit Plan (Prior DSU Plan) was terminated by the Board of Directors in conjunction with the shareholder approval of the 2019 Plan. Awards outstanding under the Prior DSU Plan remain outstanding in accordance with and pursuant to the terms thereof. As of September 30, 2024, there were
Share-based compensation expense for each of the periods presented is as follows (in thousands):
|
Three Months Ended September 30 |
Nine Months Ended September 30 |
|||||||||||||||
|
2024 |
2023 |
2024 |
2023 |
|||||||||||||
|
Research and development |
$ | $ | $ | $ | ||||||||||||
|
General and administrative |
||||||||||||||||
|
Total share-based compensation |
$ | $ | $ | $ | ||||||||||||
We recognize share-based compensation based on the fair value of each award as estimated using the Black-Scholes option valuation model. Ultimately, the actual expense recognized over the vesting period will only be for those shares that actually vest.
A summary of option activity is as follows (in thousands, except share and per share amounts):
|
Shares Underlying Options |
Weighted Average Exercise Price Per Share |
Aggregate Intrinsic Value |
||||||||||
|
Balances at December 31, 2023 |
$ | $ | ||||||||||
|
Granted |
||||||||||||
|
Expired/cancelled |
( |
) | ||||||||||
|
Forfeited |
( |
) | ||||||||||
|
Exercised |
( |
) | ||||||||||
|
Balances at September 30, 2024 |
$ | $ | ||||||||||
Information about stock options outstanding, vested and expected to vest as of September 30, 2024, is as follows:
|
Outstanding, Vested and Expected to Vest |
Options Vested and Exercisable |
||||||||||||||||||||||||||
|
Per Share Exercise Price |
Shares |
Weighted Average Remaining Contractual Life (Years) |
Weighted Average Exercise Price |
Options Exercisable |
Weighted Average Remaining Contractual Life (Years) |
||||||||||||||||||||||
| $ |
- | $ |
$ | ||||||||||||||||||||||||
| $ |
- | $ |
|||||||||||||||||||||||||
| $ |
- | $ |
|||||||||||||||||||||||||
| $ |
- | $ |
|||||||||||||||||||||||||
|
$
|
- | $ |
|||||||||||||||||||||||||
| $ | |||||||||||||||||||||||||||
ITEM 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations is based upon accounting principles generally accepted in the United States of America and discusses the financial condition and results of operations for DiaMedica Therapeutics Inc. and our subsidiaries for the three and nine months ended September 30, 2024 and 2023.
This discussion should be read in conjunction with our condensed consolidated financial statements and related notes included elsewhere in this report and our annual report on Form 10-K for the year ended December 31, 2023. The following discussion contains forward-looking statements that involve numerous risks and uncertainties. Our actual results could differ materially from the forward-looking statements as a result of these risks and uncertainties. See “Cautionary Note Regarding Forward-Looking Statements” for additional cautionary information.
Business Overview
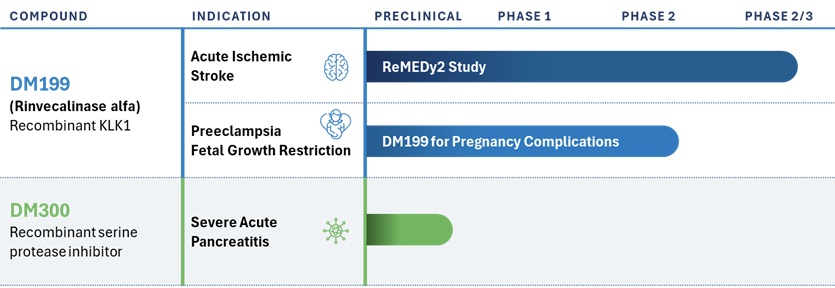
We are a clinical stage biopharmaceutical company committed to improving the lives of people suffering from severe ischemic disease with a focus on acute ischemic stroke (AIS) and preeclampsia (PE). Our lead candidate DM199 (rinvecalinase alfa; rhKLK1) is the first pharmaceutically active recombinant (synthetic) form of the human tissue kallikrein-1 (KLK1) protein (serine protease enzyme) to be clinically studied in patients. KLK1 is an established therapeutic modality in Asia, with human urinary KLK1, for the treatment of AIS and porcine KLK1 for the treatment of cardio renal disease, including hypertension. We have also produced a potential novel treatment for severe acute pancreatitis, DM300, which is currently in the early preclinical stage of development. Our long-term goal is to use our patented and in-licensed technologies to establish our Company as a leader in the development and commercialization of therapeutic treatments from novel recombinant proteins. Our current focus is on the treatment of AIS and PE. We plan to advance DM199 through required clinical trials to create shareholder value by establishing its clinical and commercial potential as a therapy for AIS and PE.
KLK1 is a serine protease enzyme that plays an important role in the regulation of diverse physiological processes via a molecular mechanism that increases production of nitric oxide, prostacyclin and endothelium-derived hyperpolarizing factors. In the case of AIS, DM199 is intended to enhance blood flow and boost neuronal survival in the ischemic penumbra by dilating arterioles surrounding the site of the vascular occlusion and inhibition of apoptosis (neuronal cell death) while also facilitating neuronal remodeling through the promotion of angiogenesis. In preeclampsia, DM199 is intended to lower blood pressure, enhance endothelial health and increase perfusion to maternal organs and the placenta, potentially disease modifying outcomes improving both maternal and perinatal outcomes.
Our product development pipeline is as follows:
AIS Phase 2/3 ReMEDy2 Trial
We are currently conducting our ReMEDy2 clinical trial of DM199 for the treatment of AIS. Our ReMEDy2 clinical trial is a Phase 2/3, adaptive design, randomized, double-blind, placebo-controlled trial intended to enroll approximately 350 patients at up to 100 sites globally. The adaptive design component includes an interim analysis by our independent data safety monitoring board after the first 200 participants. Based on the results, the study may be stopped for futility, or a new total sample size will be determined, ranging between 300 and 728 patients, according to a pre-determined statistical plan. Patients enrolled in the trial will be treated with either DM199 or placebo within 24 hours of the onset of AIS symptoms. The trial excludes patients who received mechanical thrombectomy (MT) or participants with large vessel occlusions in the intracranial carotid artery or the M1 segment for the middle cerebral, vertebral or basilary arteries or those that are otherwise eligible for MT. As a result of our recent protocol amendment, participants treated with tissue plasminogen activator (tPA) or tenecteplase (TNK), (thrombolytic agents) intended to dissolve blood clots, are now eligible for participation if they continue to experience a persistent neurological deficit after receiving thrombolytic treatment and meet all other trial criteria, including repeat brain imaging to assess any hemorrhagic (bleeding) transformation. The study population is representative of the approximately 80% of AIS patients who do not have treatment options today, primarily due to the limitations on treatment with tPA/TNK and/or MT. The primary endpoint of the ReMEDy2 trial is physical recovery from stroke as measured by the well-established modified Rankin Scale (mRS) at day 90, specifically recovering to an mRS score of 0-1 (mRS range of 0-6). We believe that our ReMEDy2 trial has the potential to serve as a pivotal registration study of DM199 in this patient population.
Prior to the clinical hold of our ReMEDy2 trial, announced in July 2022 and fully lifted in June 2023, we had experienced and are continuing to experience slower than expected site activations and enrollment in our ReMEDy2 trial. We believe these conditions may be due to hospital and medical facility staffing shortages; inclusion/exclusion criteria in the study protocol; concerns managing logistics and protocol compliance for participants discharged from the hospital to another hospital or an intermediate care facility; concerns regarding the prior clinically significant hypotension events and circumstances surrounding the previous clinical hold; and competition for research staff and trial subjects due to other pending stroke and neurological trials. We continue to reach out to current and potential study sites to understand the specific issues at each study site. In an effort to mitigate the impact of these factors, we have significantly expanded our internal clinical team and have brought in-house certain trial activities, including identification, qualification and activation, clinical site monitoring and overall program management. In addition, we made the decision to globally expand the trial and to this end we have submitted or are in the process of preparing regulatory filings and identifying and engaging study sites in the countries of Canada, Australia and Georgia. We also recently revised the study protocol to widen the inclusion criteria and reduce the burden on participants and sites. We continue to work closely with our contract research organizations and other advisors to develop procedures to support both U.S. and global study sites and potential participants as needed. We intend to continue to monitor the results of these efforts and, if necessary, implement additional actions to mitigate the impact of these factors on our ReMEDy2 trial; however, no assurances can be provided as to the success of these mitigation actions and if or when these issues will resolve. The failure to resolve these issues will result in delays in our ReMEDy2 trial.
Preeclampsia Program
On October 9, 2024, we announced the receipt of regulatory approval from SAHPRA to initiate an investigator-sponsored study of DM199 in PE. PE is a serious pregnancy disorder that typically develops after the 20th week of gestation, characterized by high blood pressure and damage to organ systems, often the kidneys and liver. Affecting up to 8% of pregnancies worldwide, preeclampsia can pose significant risks to both the mother and baby, including risk of stroke, placental abruption, progression to eclampsia, premature delivery, and death. Symptoms may include severe headaches, vision changes, upper abdominal pain and swelling in the hands and face. Delivery of the baby, often very prematurely, is the only available option for stopping the progression of preeclampsia. Women who have had preeclampsia have three to four times the risk of high blood pressure and double the risk for heart disease and stroke and there are currently no approved therapeutics for PE in the United States or Europe.
We believe DM199 has the potential to lower blood pressure, enhance endothelial health, and improve perfusion to maternal organs and the placenta. We have completed studies on fertility, embryofetal development and pre- and post-natal development in animal models, which support the potential safety in pregnant humans. Additionally, we completed a placental transfer study in pregnant rodents in which DM199 did not cross the placental barrier. Specifically, DM199 was detectable in the maternal blood, but undetectable in the fetal blood.
We are financially supporting the conduct of a Phase 2 open-label, single center, single-arm, safety and pharmacodynamic, proof-of-concept, investigator-sponsored study of DM199 for the treatment of PE at the Tygerberg Hospital, Cape Town, South Africa. Up to 90 women with PE, and potentially an additional 30 subjects with fetal growth restriction, may be evaluated. The first subject was enrolled in the fourth quarter of 2024. Participants in Part 1A of the study will include late stage PE (>34 weeks gestational age) patients. Topline data from Part 1A is expected to demonstrate initial proof-of-concept including whether DM199 is safe and lowers blood pressure. Additionally, for patients with early onset PE, we will be looking for improvements in uterine artery dilation, an indication that DM199 therapy is potentially disease modifying in these patients.
Financial Overview
We have not generated any revenues from product sales. Since our inception, we have financed our operations from public and private sales of equity, the exercise of warrants and stock options, interest income on funds available for investment and government grants. Our June 2024 private placement generated $11.7 million in net proceeds after deducting offering expenses. We have incurred losses in each year since our inception. Our net losses were $16.5 million and $14.2 million for the nine months ended September 30, 2024 and 2023, respectively. As of September 30, 2024, we had an accumulated deficit of $132.1 million. Substantially all of our operating losses resulted from expenses incurred in connection with our product candidate development programs, our research and development (R&D) activities, and general and administrative (G&A) support costs associated with our operations and status as a publicly listed company.
We expect to continue to incur significant expenses and increased operating losses for at least the next several years. We anticipate that our quarterly expenses will increase moderately relative to recent prior periods as we expand our ReMEDy2 trial globally and as enrollment continues and as we expand our DM199 clinical development program into PE and support the Phase 2 PE trial. Our efforts to expand our team to provide support for our operations and maintaining, expanding and protecting our intellectual property portfolio will also likely contribute to such increases.
While we expect our rate of future negative cash flow per month will generally increase moderately relative to recent prior periods as we continue our ReMEDy2 trial, including our global expansion, and expand our DM199 clinical development program into PE, we expect our current cash resources will be sufficient to allow us to continue our ReMEDy2 trial, support the Phase 2 PE trial and otherwise fund our planned operations for at least the next 12 months from the date of issuance of the condensed consolidated financial statements included in this report. The amount and timing of our future funding requirements will depend on many factors, including timing and results of our ongoing development efforts, including our current ReMEDy2 trial and in particular the rate of site activation and participant enrollment in the study, the Phase 2 PE trial, the potential further expansion of our current development programs and other factors. We may require or otherwise seek significant additional funds earlier than we currently expect. We may elect to raise additional funds even before we need them if market conditions for raising additional capital are favorable.
Components of Our Results of Operations
Research and Development Expenses
R&D costs include expenses incurred in the conduct of human clinical trials such as fees paid to external service providers including contract research organizations; clinical support services; clinical development including clinical site costs; outside nursing services; and laboratory testing. R&D costs also include non-clinical research studies; fees paid to contract manufacturing and development organizations and outside laboratories for the development of DM199 and related manufacturing processes; costs to produce sufficient amounts of the DM199 compound for use in our clinical studies; consulting resources with specialized expertise related to execution of our development plan for our DM199 product candidate; and personnel costs, including salaries, benefits and share-based compensation.
At this time, due to the risks inherent in the clinical development process and the clinical stage of our product development programs, we are unable to estimate with any certainty the costs we will incur in developing DM199 through marketing approval or any of our preclinical development programs. The process of conducting clinical studies necessary to obtain regulatory approval and manufacturing scale-up to support expanded development and potential future commercialization is costly and time consuming. Any failure by us or delay in completing clinical studies, manufacturing scale-up or in obtaining regulatory approvals could lead to increased R&D expenses and, in turn, have a material adverse effect on our results of operations.
We expect that our R&D expenses will increase moderately in the future relative to recent prior periods if we are successful in advancing DM199, or any of our preclinical programs, through the required stages of clinical development. The process of conducting clinical trials necessary to obtain regulatory approval and manufacturing scale-up to support expanded development and potential future commercialization is costly and time consuming.
General and Administrative Expenses
G&A expenses consist primarily of salaries, employee benefits, share-based compensation and other personnel costs related to our executive, finance, business development and support functions. G&A expenses also include insurance, including directors and officers liability coverage, rent and utilities, travel expenses, patent costs, and professional fees, including for auditing, tax and legal services.
Other Income, Net
Other income, net consists primarily of interest income earned on marketable securities.
Results of Operations
Comparison of the Three and Nine Months Ended September 30, 2024 and 2023
The following table summarizes our unaudited results of operations for the three and nine months ended September 30, 2024 and 2023 (in thousands):
|
Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
|
2024 |
2023 |
2024 |
2023 |
|||||||||||||
|
Research and development expenses |
$ | 4,983 | $ | 3,272 | $ | 12,587 | $ | 9,433 | ||||||||
|
General and administrative expenses |
1,900 | 1,885 | 5,675 | 5,986 | ||||||||||||
|
Other income, net |
(616 | ) | (693 | ) | (1,739 | ) | (1,220 | ) | ||||||||
Research and Development Expenses
R&D expenses increased to $5.0 million for the three months ended September 30, 2024, up from $3.3 million for the three months ended September 30, 2023. R&D expenses increased to $12.6 million for the nine months ended September 30, 2024, up from $9.4 million for the nine months ended September 30, 2023. These increases are due primarily to cost increases resulting from the continuation of our ReMEDy2 clinical trial, the expansion of our clinical team and increased manufacturing development activity. These increases were partially offset by cost reductions related to clinical trial work completed in 2023, including our Phase 1C and REDUX trials, and the completion in 2023 of in-use study work performed to address the clinical hold on our ReMEDy2 trial. We expect our R&D expenses to increase moderately relative to recent prior periods as we globally expand the ReMEDy2 trial site activations and participant enrollments continue.
General and Administrative Expenses
G&A expenses were $1.9 million for each of the three months ended September 30, 2024 and September 30, 2023. G&A expenses were $5.7 million for the nine months ended September 30, 2024, down from $6.0 million for the nine months ended September 30, 2023. The decrease for the nine-month comparison resulted from the combination of reductions in the cost of directors and officers liability insurance premiums and decreased legal fees incurred in connection with our lawsuit against PRA Netherlands and was partially offset by increased personnel costs incurred in conjunction with expanding our team and increased non-cash share-based compensation costs. We expect G&A expenses to remain steady as compared to prior periods.
Other Income, Net
Other income, net was $616 thousand and $1.7 million for the three and nine months ended September 30, 2024, respectively, compared to $693 thousand and $1.2 million for the three and nine months ended September 30, 2023, respectively. The increase for the nine-month comparison was driven by increased interest income recognized during the current year periods related to higher marketable securities balances during the current year periods as compared to the same periods in the prior year.
Liquidity and Capital Resources
The following tables summarize our liquidity and capital resources as of September 30, 2024 and December 31, 2023, and our cash flows for each of the nine month periods ended September 30, 2024 and 2023, and are intended to supplement the more detailed discussion that follows (in thousands):
|
September 30, 2024 |
December 31, 2023 |
|||||||
|
Cash, cash equivalents and marketable securities |
$ | 50,197 | $ | 52,895 | ||||
|
Total assets |
52,524 | 54,160 | ||||||
|
Total current liabilities |
4,297 | 2,786 | ||||||
|
Total shareholders’ equity |
47,964 | 51,057 | ||||||
|
Working capital |
46,470 | 50,889 | ||||||
|
Nine Months Ended September 30, |
||||||||
|
2024 |
2023 |
|||||||
|
Cash Flow Data |
||||||||
|
Cash flow provided by (used in): |
||||||||
|
Operating activities |
$ | (15,642 | ) | $ | (14,916 | ) | ||
|
Investing activities |
3,359 | (24,423 | ) | |||||
|
Financing activities |
11,874 | 36,843 | ||||||
|
Net decrease in cash and cash equivalents |
$ | (409 | ) | $ | (2,496 | ) | ||
Working Capital
We had aggregate cash, cash equivalents and marketable securities of $50.2 million, current liabilities of $4.3 million and working capital of $46.5 million as of September 30, 2024, compared to aggregate cash, cash equivalents and marketable securities of $52.9 million, $2.8 million in current liabilities and $50.9 million in working capital as of December 31, 2023. The decreases in our combined cash, cash equivalents and marketable securities and in our working capital are due to the net cash used in operating activities partially offset by the net proceeds received from our June 2024 private placement.
Cash Flows
Operating Activities
Net cash used in operating activities was $15.6 million for the nine months ended September 30, 2024 compared to $14.9 million for the nine months ended September 30, 2023. The increase in cash used in operating activities resulted primarily from the combination of increased net loss and the advance of deposit funds to vendors supporting our ReMEDy2 clinical trial during the current year period, partially offset by changes in operating assets and liabilities during the current year period.
Investing Activities
Investing activities consist primarily of purchases and maturities of marketable securities. Net cash provided by investing activities was $3.4 million for the nine months ended September 30, 2024 compared to net cash used in investing activities of $24.4 million for the nine months ended September 30, 2023. This change resulted primarily from the timing of maturities and investments in marketable securities.
Financing Activities
Net cash provided by financing activities was $11.9 million and $36.8 million for the nine months ended September 30, 2024 and 2023, respectively, and for each period was comprised principally of net proceeds from the sale of common shares in private placement offerings.
Capital Requirements
Since our inception, we have incurred losses while advancing the R&D of our DM199 product candidate. We have not generated any revenues from product sales and do not expect to do so for at least three to four years. We do not know when or if we will generate any revenues from product sales or out-licensing of our DM199 product candidate or any future product candidate. We do not expect to generate any revenue from product sales unless and until we obtain required regulatory approvals. We expect to continue to incur substantial operating losses until such time as any future product sales, licensing fees, milestone payments and/or royalty payments are sufficient to generate revenues to fund our continuing operations. We expect our operating losses to increase as compared to prior periods as we continue the research, development and clinical studies of, and seek regulatory approval for, our DM199 product candidate, including, in particular, the continuation and global expansion of our ReMEDy2 trial. In the long-term, subject to obtaining regulatory approval of our DM199 product candidate, or any future product candidate, and in the absence of the assistance of a strategic partner, we expect to incur significant commercialization expenses for product sales, marketing, manufacturing and distribution.
Accordingly, we expect we will need substantial additional capital to further our R&D activities, current and potential future clinical studies, regulatory activities and otherwise develop our product candidate, DM199, or any future product candidate, to a point where the product candidate may be out-licensed or commercially sold. Although we are striving to achieve these plans, there is no assurance that these and other strategies will be achieved or that additional funding will be obtained on favorable terms or at all. We expect our rate of future negative cash flow per month will vary depending on our clinical activities and the timing of expenses incurred and will increase moderately relative to recent prior periods as we continue and globally expand our ReMEDy2 trial and the Phase 2 PE trial is conducted. We expect our current cash resources will be sufficient to continue our ReMEDy2 trial, support the Phase 2 PE trial and otherwise fund our planned operations for at least the next twelve months from the date of issuance of the condensed consolidated financial statements included in this report. The amount and timing of our future funding requirements will depend on many factors, including timing and results of our ongoing development efforts, including our current ReMEDy2 trial and the Phase 2 PE trial, the potential further expansion of our current development programs and other factors on our operating expenses. We may require significant additional funds earlier than we currently expect and there is no assurance that we will not need or seek additional funding prior to such time, especially if market conditions for raising additional capital are favorable.
Historically, we have financed our operations primarily from sales of equity securities and the exercise of warrants and stock options, and we expect to continue this practice for the foreseeable future. We do not have any existing credit facilities under which we could borrow funds. We may seek to raise additional funds through various sources, such as equity or debt financings, or through strategic collaborations and license agreements. We can give no assurances that we will be able to secure additional sources of funds to support our operations, or if such funds are available to us, that such additional financing will be sufficient to meet our needs or on terms acceptable to us. This is particularly true if our clinical data is not positive or economic and market conditions deteriorate.
To the extent we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our shareholders will be diluted. Debt financing, if available, may involve agreements that include conversion discounts, pledging our intellectual property as collateral or covenants limiting or restricting our ability to take specific actions, such as incurring additional debt or making capital expenditures. If we raise additional funds through government or other third-party funding, marketing and distribution arrangements or other collaborations, or strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. The availability of financing will be affected by the status of our clinical trials; our clinical data and other results of scientific and clinical research; the ability to obtain regulatory approvals and other regulatory actions; market acceptance of our product candidates; the state of the capital markets generally with particular reference to pharmaceutical, biotechnology and medical companies; the status of strategic alliance agreements; and other relevant commercial considerations.
If adequate funding is not available when needed, we may be required to scale back our operations by taking actions that may include, among other things, implementing cost reduction strategies, such as reducing use of outside professional service providers, reducing the number of our employees or employee compensation, modifying or delaying the development of our DM199 product candidate; licensing to third parties the rights to commercialize our DM199 product candidate for AIS, PE or other indications that we would otherwise seek to pursue, or otherwise relinquishing significant rights to our technologies, future revenue streams, research programs or product candidates or granting licenses on terms that may not be favorable to us; and/or divesting assets or ceasing operations through a merger, sale, or liquidation of our Company.
Critical Accounting Policies and Estimates
There have been no material changes to our critical accounting policies and estimates from the information provided in “Part II. Item 7, Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies,” included in our annual report on Form 10-K for the fiscal year ended December 31, 2023.
ITEM 3 QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
As a smaller reporting company, we are not required to provide disclosure pursuant to this item.
ITEM 4 CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the United States Securities Exchange Act of 1934, as amended (Exchange Act)) that are designed to provide reasonable assurance that information required to be disclosed by us in the reports we file or submit under the Exchange Act, is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. Our management evaluated, with the participation of our Chief Executive Officer and Chief Financial Officer, the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered in this report. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of the end of such period to provide reasonable assurance that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting that occurred during the three months ended September 30, 2024 that has materially affected or is reasonably likely to materially affect our internal control over financial reporting.
PART II OTHER INFORMATION
ITEM 1. LEGAL PROCEEDINGS
Litigation with Pharmaceutical Research Associates Group B.V., acquired by ICON plc as of July 1, 2021, (ICON/PRA Netherlands)
On November 23, 2022, we filed a petition requesting leave for a prejudgment attachment of all relevant documents in possession of Pharmaceutical Research Associates Group B.V., acquired by ICON plc as of July 1, 2021, (ICON/PRA Netherlands), which was granted on November 28, 2022, by the District Court of Northern Netherlands. A representative of the District Court served ICON/PRA Netherlands with the prejudgment attachment on or about December 7 and 8, 2022. The case was formally introduced to the Netherlands Commercial Court (NCC) on December 28, 2022 and a hearing by the NCC to determine whether we are entitled to take possession of the records seized was scheduled and held on March 16, 2023.
On April 21, 2023, the NCC issued a judgement affirming our ownership of the physical documents, including 51 hardcopy folders and certain digital files, related to the clinical studies performed by ICON/PRA Netherlands and seized by the Dutch courts in December 2022. The NCC further ordered ICON/PRA Netherlands to allow and tolerate the surrender of the documents, including digital and source data. Additionally, the NCC found that we are not in breach of any obligation under the clinical study agreement and ICON/PRA Netherlands had no basis to suspend the fulfillment of its obligations under the clinical study agreement to provide us all clinical data and access to perform an audit of the study. On June 15, 2023, ICON/PRA Netherlands filed an appeal of this decision and requested a scheduling hearing with the NCC, which occurred on September 23, 2024.
The hearing addressing our claims for damages was conducted on December 7, 2023. On February 7, 2024, the NCC issued a judgement in which they found that, although all data related to the study is the rightful property of DiaMedica, they found that there was an insufficient causal link between ICON/PRA Netherlands withholding study data and the damages claimed by us. We have notified the NCC and ICON/PRA Netherlands of our intent to appeal this decision and submitted our statement of grounds for appeal on October 15, 2024. The NCC has issued a decision to consolidate both appeals and evaluate them concurrently at a single hearing currently scheduled for March 2025.
From time to time, we may be subject to other various ongoing or threatened legal actions and proceedings, including those that arise in the ordinary course of business, which may include employment matters and breach of contract disputes. Such matters are subject to many uncertainties and to outcomes that are not predictable with assurance and that may not be known for extended periods of time. Other than the ICON/PRA Netherlands matter noted above, we are not currently engaged in or aware of any threatened legal actions.
ITEM 1A. RISK FACTORS
Although Item 1A. is inapplicable to us as a smaller reporting company, we hereby disclose the following new risk factors in addition to those disclosed in our annual report on Form 10-K for the fiscal year ended December 31, 2023:
The expansion of our DM199 clinical development program into PE involves certain risks related to timing, regulatory approvals, costs and enrollment. Furthermore, because the proof-of-concept clinical trial will be investigator sponsored, we will have less control over the clinical trial and may face additional risks. Any such risks could adversely impact our business and results of operations and financial position and harm our ability to raise additional financing, if and when needed.
On October 9, 2024, we announced the receipt of regulatory approval from SAHPRA to initiate an investigator-sponsored study of DM199 in PE. We are financially supporting the conduct of the Phase 2 open-label, single center, single-arm, safety and pharmacodynamic, proof-of-concept, investigator-sponsored study of DM199 for the treatment of PE at the Tygerberg Hospital, Cape Town, South Africa. We anticipate that up to 90 women with PE and potentially an additional 30 subjects with fetal growth restriction will be evaluated. The first subject was enrolled in the fourth quarter of 2024. Part 1A topline study results are intended to demonstrate initial proof-of-concept results including whether DM199 is safe, lowers blood pressure, and dilates intrauterine arteries to increase placental blood flow and are expected in the first half of 2025.
The expansion of our DM199 clinical development program into PE and the progress of that program may not occur on the anticipated timeline or at all. In addition, the Phase 2 trial may cost us more than we anticipate. Additionally, because the trial will be investigator-sponsored, we will have less control over the timing and costs of the study and the ability to recruit trial subjects than if we conducted the study with our own personnel. There is no guarantee that our physician collaborators will devote adequate time and resources to perform this study and/or maintain adequate clinical trial information regarding our product candidate. If these third parties fail to meet expected deadlines, fail to transfer to us any regulatory information in a timely manner, fail to adhere to the study protocol or fail to act in accordance with regulatory requirements or our agreement with them, or if they otherwise perform in a substandard manner or in a way that compromises the quality or accuracy of their activities or the data they obtain, then our current or future clinical trials may be extended or delayed with additional costs incurred, or our data may be rejected by applicable regulatory agencies. Any of these risks could adversely impact our business, results of operations and financial position, including our ability to raise additional financing, if and when needed.
Our drug candidate, DM199, is currently in clinical development in two areas, AIS and PE. Data from the investigator-sponsored Phase 2 PE clinical trial may adversely affect our current ReMEDy2 clinical trial, which could adversely impact our business, results of operations and financial position and harm our ability to raise additional financing, if and when needed.
We anticipate Part 1A topline study results for the Phase 2 PE clinical trial in the first half of 2025. Part 1A topline study results are intended to demonstrate whether DM199 is safe for PE patients, lowers blood pressure and/or dilates intrauterine arteries to increase placental blood flow. The results from Part 1A of the study may not be consistent with the safety results from our prior ReMEDy1 trial of DM199 for the treatment of acute ischemic stroke or certain efficacy results from that trial. If the Part 1A topline study results are inconsistent, inaccurate, incomplete or otherwise demonstrate that DM199 is not safe, does not lower blood pressure or dilate intrauterine arteries, our current ReMEDy2 clinical trial of DM199 for the treatment of acute ischemic stroke may be adversely affected. Should this occur, we may be required to repeat clinical or non-clinical studies, our clinical development plans may be significantly delayed, and we may incur additional costs, which could adversely impact our business, results of operations and financial position. Adverse results from the Part 1A topline study also could adversely affect our ability to raise additional financing, if and when needed.
ITEM 2. UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS
We did not sell any unregistered equity securities of our Company during the quarter ended September 30, 2024.
ITEM 3. DEFAULTS UPON SENIOR SECURITIES
Not applicable.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
ITEM 5. OTHER INFORMATION
Rule 10b5-1 Plan and Non-Rule 10b5-1 Trading Arrangement Adoptions, Terminations, and Modifications
During the three months ended September 30, 2024, of our directors or “officers” (as defined in Rule 16a-1(f) under the Exchange Act) adopted or terminated a “Rule 10b5-1 trading arrangement” or “non-Rule 10b5-1 trading arrangement,” as each term is defined in Item 408 of SEC Regulation S-K.
ITEM 6. EXHIBITS
The following exhibits are being filed or furnished with this quarterly report on Form 10-Q:
|
Exhibit No. |
Description |
Manner of Filing |
||
|
3.1 |
Notice of Articles of DiaMedica Therapeutics Inc. dated June 16, 2023 |
Filed herewith |
||
|
3.2 |
Amended and Restated Articles of DiaMedica Therapeutics Inc. Effective May 17, 2023 |
Incorporated by reference to Exhibit 3.1 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on May 18, 2023 (File No. 001-36291) |
||
|
31.1 |
Filed herewith |
|||
|
31.2 |
Filed herewith |
|||
|
32.1 |
Furnished herewith |
|||
|
32.2 |
Furnished herewith |
|||
|
101 |
Financial statements from the quarterly report on Form 10-Q of DiaMedica Therapeutics Inc. for the three and nine months ended September 30, 2024, formatted in Inline XBRL: (i) the Condensed Consolidated Balance Sheets, (ii) Condensed Consolidated Statements of Operations and Comprehensive Loss, (iii) Condensed Consolidated Statements of Shareholders’ Equity, (iv) Condensed Consolidated Statements of Cash Flows, and (v) Notes to the Condensed Consolidated Financial Statements. |
Filed herewith |
||
|
104 |
Cover Page Interactive Data File |
Embedded within the Inline XBRL document |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
|
DIAMEDICA THERAPEUTICS INC. |
|
|
Date: November 13, 2024 |
/s/ Rick Pauls |
|
Rick Pauls |
|
|
President and Chief Executive Officer |
|
|
(Principal Executive Officer) |
|
|
Date: November 13, 2024 |
/s/ Scott Kellen |
|
Scott Kellen |
|
|
Chief Financial Officer |
|
|
(Principal Financial Officer and Principal Accounting Officer) |